
Die klinische Bewertung von Studien erfolgt künftig auf europäischer Ebene. Die Beurteilung des Zusatznutzens und die Preisgestaltung verbleiben weiterhin in der nationalen Zuständigkeit. (Foto von Guillaume Périgois auf Unsplash)
Das Ziel ist es, durch eine gemeinsame klinische Bewertung neue Medikamente effizienter und EU-weit einheitlich zu bewerten. Die erste Phase umfasst Arzneimittel gegen Krebserkrankungen und neuartige Therapieansätze (ATMPs). Ab 2028 folgen Orphan Drugs, und bis 2030 sollen alle Arzneimittel mit neuen Wirkstoffen einbezogen werden.
Die europäische Zusammenarbeit, basierend auf der Verordnung 2021/2282 des EU-Parlaments, könnte den Aufwand nationaler Bewertungsverfahren erheblich reduzieren. Experten wie Prof. Dieter Cassel und Prof. Volker Ulrich betonen, dass die Praxis zeigen muss, ob diese ambitionierte Struktur für die 27 Mitgliedstaaten effektiv funktioniert.
Der BPI und Pharma Deutschland sehen insbesondere bei innovativen Therapieansätzen wie ATMPs einige Herausforderungen auf Deutschland zukommen. Und vor allem vor diesem Hintergrund fordern Experten wie Cassel und Ulrich neue Bewertungs- und Vergütungsmodelle. Pay-for-Performance-Modelle könnten die bestehenden Verfahren sinnvoll ergänzen. Vorwiegend bei Orphan Drugs, deren Nutzen oft nicht durch klassische Studien nachweisbar ist, müssen die methodischen Anforderungen stärker auf die Therapiesituation abgestimmt werden, betont der BPI.
Konkret müssen die Mitgliedstaaten den europäischen HTA-Bericht bei ihren Entscheidungen angemessen berücksichtigen, dürfen aber bei Bedarf klinische Zusatzanalysen einfordern. Ein verbindlicher Mechanismus regelt die einmalige Einreichung klinischer Daten auf EU-Ebene, die auf nationaler Ebene nicht erneut angefragt und eingereicht werden dürfen.
Nationale Gesundheitsbehörden müssen europäisch handeln
Pharma Deutschland gibt zu bedenken, dass es bislang keine klaren europäischen Beratungsmöglichkeiten gibt und unklar bleibt, wie die Ergebnisse der EU-HTA in nationale Bewertungsverfahren integriert werden sollen.
Dorothee Brakmann, Hauptgeschäftsführerin von Pharma Deutschland, meint zu diesem Punkt: „Zwar hat das Bundesministerium für Gesundheit Vorschläge zur Änderung der Arzneimittelnutzenverordnung eingebracht, die den europäischen Prozess besser mit dem nationalen AMNOG-Verfahren verzahnen sollen. Dennoch fehlt es weiterhin an klaren Regelungen für die inhaltliche Abstimmung.“
Der vfa findet, dass „es ergänzender nationaler Regelungen bedarf, die eine nahtlose Verbindung zum europäischen Prozess schaffen. Gelingt diese Integration, kann das EU-HTA den Pharmastandort Deutschland und Europa stärken.“
Der Ablauf des europäischen Nutzenbewertungsverfahrens in fünf Phasen:
Initiierungsphase
- Information des HTA – Sekretariats über die Planung eines Zulassungsantrages, Versand des „Letter of Intent“ (Absichtserklärung einer Beantragung einer Zulassung) an die European Medicines Agency (EMA) und das HTA-Sekretariat inklusive aller erforderlichen Informationen
- Bestimmung der Gutachter seitens der HTA-Koordinierungsgruppe / Untergruppe und Identifikation von Patienten und klinischen Experten
- Veröffentlichung der Information über den Beginn des Verfahrens
Scoping Phase
- Einbindung der Patienten und klinischen Experten
- Übermittlung der Fragestellungen (PICO1) der Mitgliedsstaaten für das JCA
- Konsolidierung der Fragestellung
- Information des Herstellers über die im JCA zu beantwortenden Fragestellungen und Aufforderung zur Einreichung eines Dossiers
Dossiereinreichungsphase
- Erstellung des Dossiers anhand der Dossiervorgaben und methodischer Leitfäden
- Einreichung des Dossiers innerhalb der vorgegebenen Fristen (gemäß HTAR und Durchführungsrechtsakt für JCA mindestens 45 Tage vor der Positive Opinion des Ausschusses für Humanarzneimittel derEMA)
- Vollständigkeitsprüfung und Information über fehlende Informationen
- Ggf. Nachreichung fehlender Informationen
- Ggf. Aktualisierung des Dossiers im Falle von Anwendungsgebietsänderungen oder neuen Daten, die im Rahmen des Zulassungsprozesses bekannt werden.
Bewertungsphase
- Bewertung des Dossiers durch die Gutachter
- Ggf. Rückfragen während der Bewertung an den pharmazeutischen Unternehmer
- Zusendung des HTA - Berichtentwurfes an den pharmazeutischen Unternehmer zum Zwecke der Faktenüberprüfung
- Abnahme des finalen HTA-Berichtes durch die HTA – Koordinierungsgruppe
Veröffentlichung
- Veröffentlichung des finalen HTA-Berichtes und des Dossiers ohne Betriebs- und Geschäftsgeheimnisse (Billigung des HTA-Berichts durch die Koordinierungsgruppe 30 Tage nach Entscheidung der EU-Kommission über die Zulassung + 10 Tage verfahrenstechnische Prüfung durch die EU-Kommission = 40 Tage nach Zulassung).
- Der Bericht darf keine Werturteile enthalten. Die Schlussfolgerungen, die die nationalen Gesundheitssysteme aus dem Bericht ziehen, obliegen weiterhin der Verantwortung der Mitgliedstaaten.
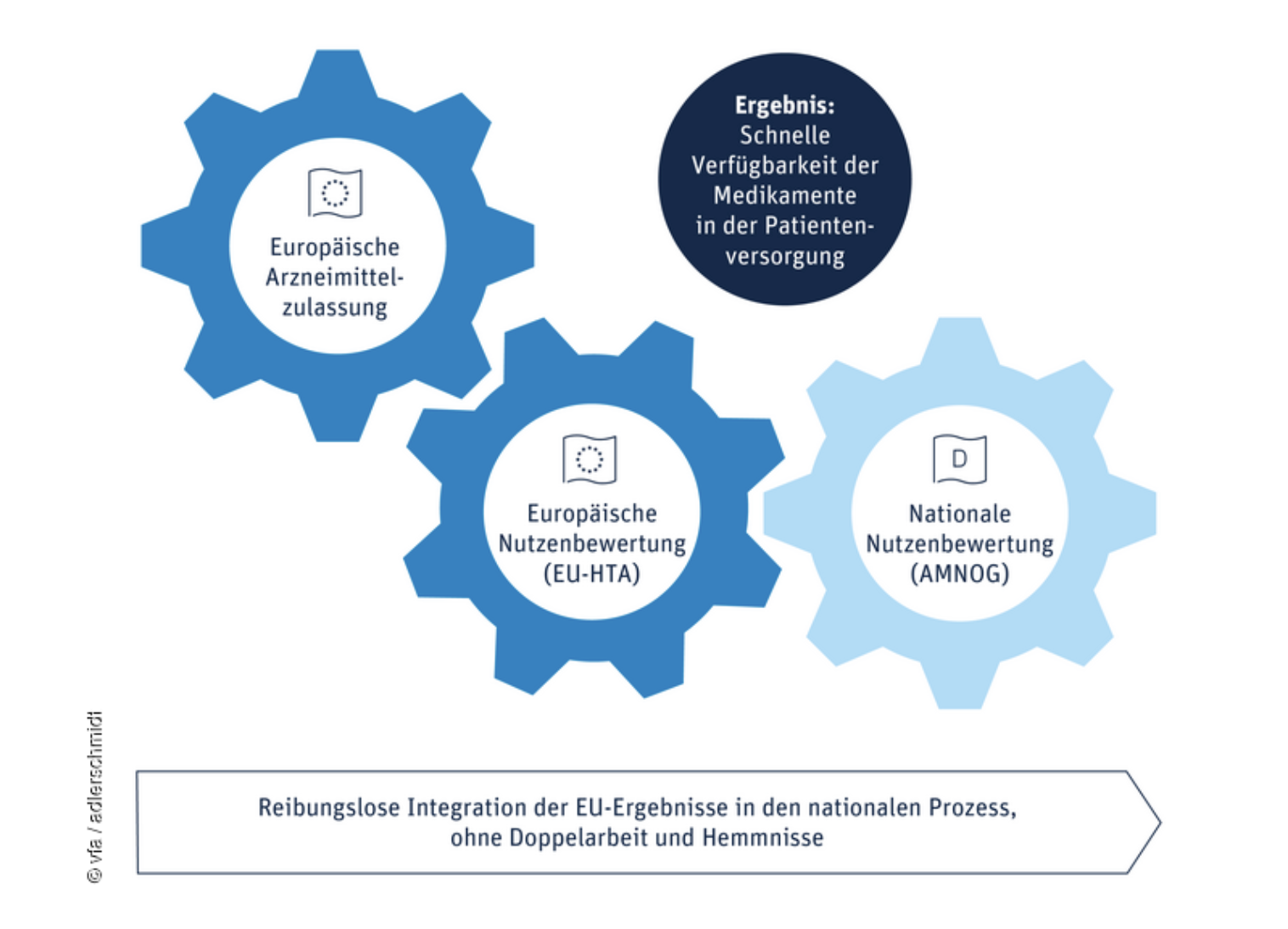
Grafik: vfa
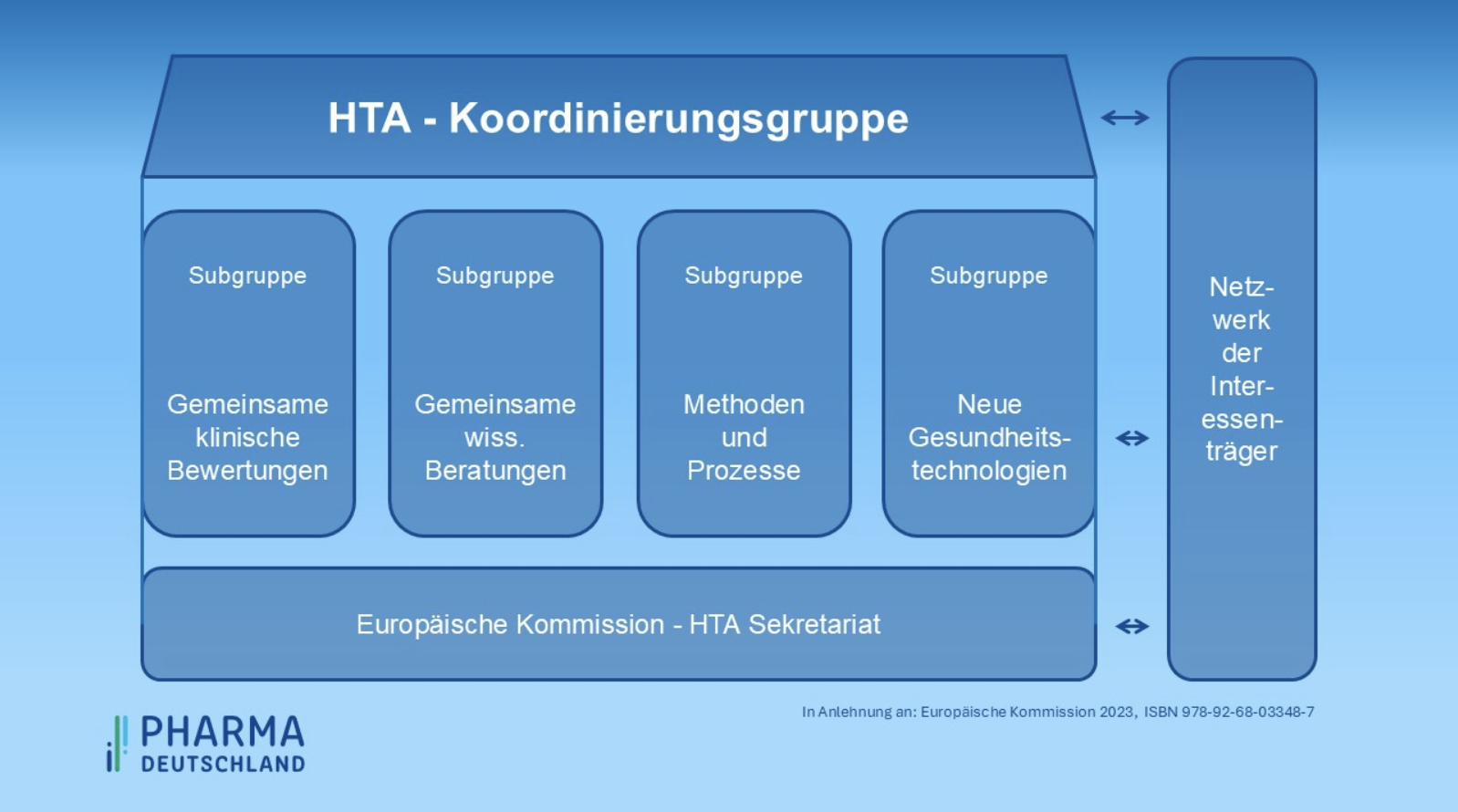
Bild: Pharma Deutschland
Kernergebnisse der BPI-AMNOG-Daten 2024:
- Rund die Hälfte aller Erstbewertungsverfahren ohne Zusatznutzen – oftmals aus methodischen beziehungsweise formalen Gründen: In einer Vielzahl der Fälle konnte aus methodischen Gründen kein Zusatznutzen festgestellt werden. Die vorliegenden Daten wurden gar nicht ausgewertet.
- Insgesamt wurden 922 Nutzenbewertungsverfahren in Deutschland bis Ende 2023 rechtskräftig abgeschlossen, darunter 802 Erstbewertungen und 120 erneute Bewertungen.
- Bei den Erstbewertungen wurde dem jeweiligen Wirkstoff in gut der Hälfte aller Verfahren (51,1%) ein Zusatznutzen anerkannt – rund die Hälfte der geprüften Präparate bekam keinen Zusatznutzen attestiert (48,9%).
- 49 Marktaustritte: Bis Ende 2023 wurden nach erfolgter Zulassung insgesamt 49 Therapien wieder vom Markt zurückgezogen – darunter 45 nach Abschluss des Preisfindungsprozesses (AMNOG-Verfahrens).
- Gute Verfügbarkeit von Innovationen in Deutschland: 88% der zwischen 2019 und 2022 in der EU eingeführten Arzneimittel sind in Deutschland verfügbar.
- Auch bei der durchschnittlichen Zeit bis zur Erstattung eines Arzneimittels ist Deutschland mit 126 Tage schnell (EU-Durchschnitt liegt bei 517 Tagen).
Erhalten Sie jetzt uneingeschränkten Zugriff auf alle interessanten Artikel.
- Online-Zugriff auf das PM-Report Heftarchiv
- Aktuelle News zu Gesundheitspolitik, Pharmamarketing und alle relevanten Themen
- 11 Ausgaben des PM-Report pro Jahr inkl. Specials
