
Im Editorial des G-BA-Geschäftsberichts wird u.a. betont: „Bei aller Verbesserung im Detail war das zurückliegende Jahr 2024 nicht einfach. Weiter verschärfte sich der Kostendruck durch steigende Gesundheitsausgaben und den wachsenden medizinischen Bedarf einer alternden Gesellschaft mit vielen Alleinlebenden – dies alles bei anhaltendem Fachkräftemangel. Zugleich war das Jahr geprägt von der Innovationskraft in Forschung, Wissenschaft und Medizin, was jedoch wieder Kostensteigerungen mit sich bringt. Auf diese Herausforderungen weise zu reagieren und die Gesundheitsversorgung zukunftsfest aufzustellen, war und bleibt eine Herkulesaufgabe.“ (Foto von Diana Polekhina auf Unsplash)
Der Gemeinsame Bundesausschuss (G-BA) legte auch 2024 ein hohes Arbeitstempo an den Tag: Mit 693 Einzelbeschlüssen wurde das Vorjahresniveau leicht übertroffen. Trotz schwieriger Rahmenbedingungen (steigende Gesundheitskosten, Fachkräftemangel) konnte die Versorgung gesetzlich Versicherter weiterentwickelt werden. Thematische Schwerpunkte lagen auf Kindergesundheit, Digitalisierung, Versorgungsqualität und Arzneimittelbewertung.
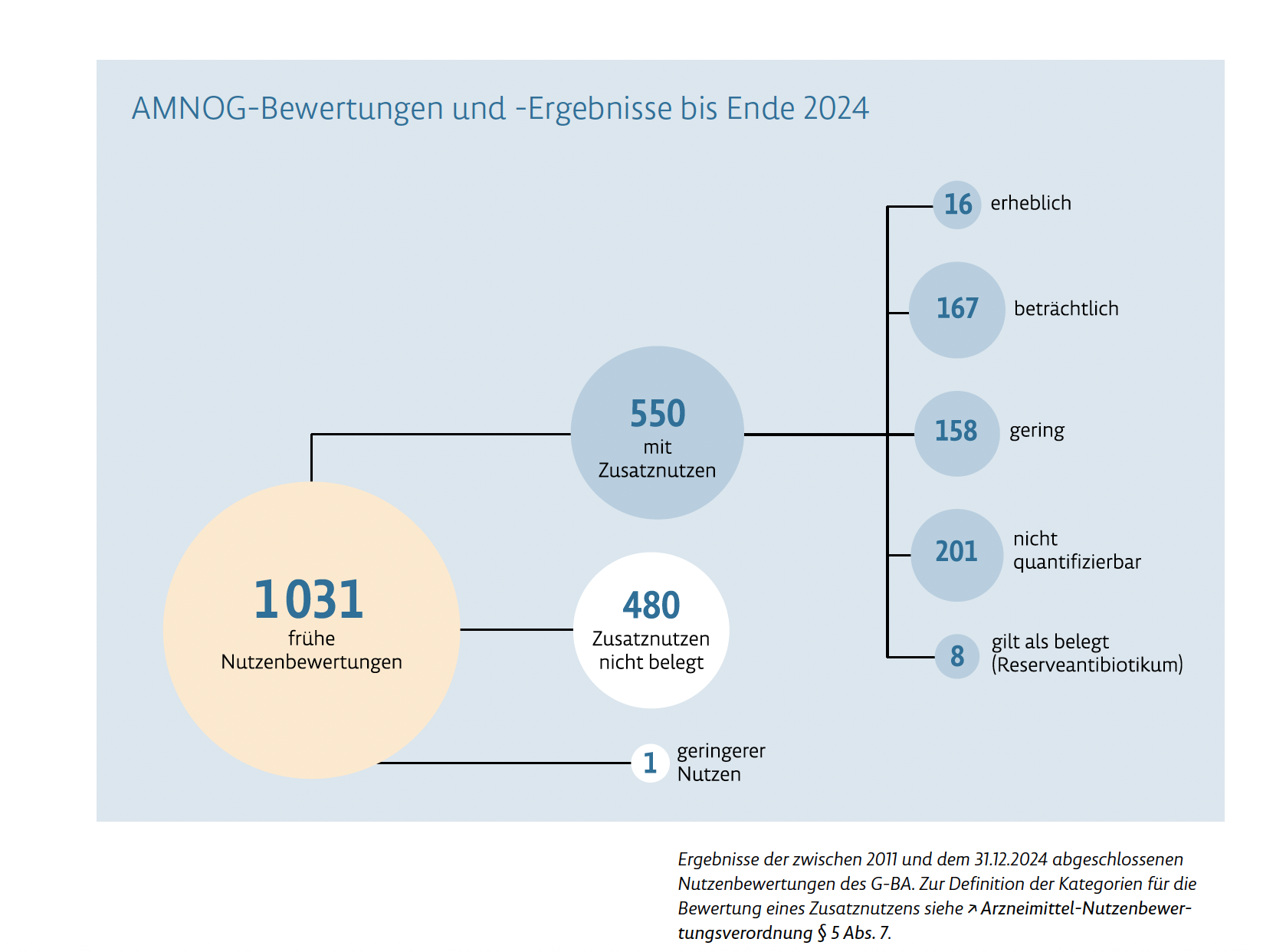
Fokus 1: AMNOG-Bewertungen – Orphan Drugs im Zusatznutzen-Check
Seit Inkrafttreten des AMNOG (Arzneimittelmarktneuordnungsgesetz) im Jahr 2011 bewertet der G-BA alle neu zugelassenen Arzneimittel hinsichtlich ihres Zusatznutzens gegenüber der Standardtherapie. Die Bewertungen sind Grundlage für spätere Preisverhandlungen mit den Krankenkassen.
Zahlen & Fakten:
- 540 Wirkstoffe wurden bis Ende 2024 bewertet.
- Etwa 20% der Bewertungen betreffen sogenannte Orphan Drugs (Arzneimittel gegen seltene Erkrankungen).
- 53 % der bewerteten Wirkstoffe zeigten einen Zusatznutzen.
- Nur 16 davon wiesen einen erheblichen Zusatznutzen auf.
Orphan-Drug-Sonderregelung:
Orphan Drugs genießen laut Gesetz ein „Orphan-Privileg“: Ihr Zusatznutzen gilt bis zu einem Umsatz von 30 Mio. Euro pro Jahr automatisch als belegt. Erst bei Überschreiten dieser Schwelle erfolgt eine vollständige Nutzenbewertung – mit Vergleich zur zweckmäßigen Standardtherapie.
Erkenntnisse 2024:
- 23 Orphan Drugs wurden 2024 nachbewertet.
- In 18 Fällen führte dies zu einem niedrigeren Preis, weil der zuvor angenommene Zusatznutzen nicht bestätigt werden konnte.
- Nur 5 Wirkstoffe bestätigten ihre ursprüngliche Zusatznutzen-Kategorie.
- Das zeigt laut G-BA: Vollständige Bewertungen sind entscheidend, um therapeutischen Nutzen realistisch einzuschätzen und Preis-Leistungsverhältnisse zu wahren.
Bewertung durch das IQWiG:
Ein begleitendes Arbeitspapier des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) analysierte erstmals systematisch den Einfluss von Nachbewertungen auf Erstattungspreise. Das Ergebnis spricht klar für eine striktere Prüfung von Orphan Drugs nach Überschreiten der Umsatzgrenze.
Fokus 2: Anwendungsbegleitende Studien – Erste Erfahrungen
Seit 2020 kann der G-BA bei Arzneimitteln mit unzureichender Datenlage zum Markteintritt sogenannte Anwendungsbegleitende Datenerhebungen (AbD) anordnen – meist in Form von Registerstudien. Ziel ist es, Evidenzlücken zu schließen und eine spätere belastbare Nutzenbewertung zu ermöglichen.
Umsetzung 2024:
- Der G-BA traf 25 AbD-Beschlüsse.
- Insgesamt fünf Registerstudien laufen bereits.
- Die erste bundesweite AbD-Registerstudie zum Gentherapeutikum Zolgensma startete 2022. Die erneute Nutzenbewertung ist für 2027 vorgesehen.
G-BA-Strategie:
- Der G-BA betreibt ein Monitoring der EMA-Zulassungsanträge, um frühzeitig zu erkennen, wann eine AbD erforderlich wird.
- Die Vorbereitungen für eine Registerstudie beginnen idealerweise parallel zur Zulassung.
- Ziel: Alle behandelten Patient:innen sollen direkt nach Markteintritt in die AbD einbezogen werden.
Herausforderungen und Entwicklungsbedarf:
- In der Praxis zeigt sich: Die Umsetzung ist aufwendig und strukturell noch nicht effizient.
- Kritikpunkt: Die gesetzliche Regelung ermöglicht bisher keinen Zwang zur Register-Standardisierung oder zum Teilen bestehender Studienunterlagen.
G-BA fordert:
- Verpflichtende Bereitstellung von Herstellerdaten gegen Entschädigung.
- Zentrale Registerstrukturen, die auch mehrere Wirkstoffe in einer Indikation abdecken.
- Einbindung der Zulassungsbehörden und Fachgesellschaften zur Etablierung standardisierter Register vor Markteintritt.
Prof. Josef Hecken, Unparteiischer G-BA-Vorsitzender, kritisiert dementsprechend: „Das Instrument ist in seiner derzeitigen gesetzgeberischen Ausgestaltung nicht effizient und sollte nachgebessert werden.“
Grafik: G-BA
Erhalten Sie jetzt uneingeschränkten Zugriff auf alle interessanten Artikel.
- Online-Zugriff auf das PM-Report Heftarchiv
- Aktuelle News zu Gesundheitspolitik, Pharmamarketing und alle relevanten Themen
- 11 Ausgaben des PM-Report pro Jahr inkl. Specials
